
企业名称:郑州永信医疗器械有限公司
联系人:李经理
手机: 15003718881 15003718887 15803716767
监督:15003716161
邮箱:58775877@qq.com
网址:www.yongxinyiliao.com
地址:郑州高新技术产业开发区莲花街316号5幢20层88号
日前,科技部布了《科技部办公厅关于优化人类遗传资源行政审批流程的通知》国科办社函[2017]717号。
通知表示,科技部研究制定了针对为获得相关药物和医疗器械在我国的上市许可,利用我国人类遗传资源展开国际合作临床试验的优化审批流程。
主要优化内容主要包括:
鼓励多中心临床研究设立组长单位,一次性申报;
临床试验成员单位认可组长单位的伦理审查,不再重复申报;
具有法人资格的合作双方共同申请;
调整提交伦理批件、临床试验批件的时间,由原来的在线申报时提交延后至正式受理时提交;
取消省级科技行政部门和国务院有关部门科技主管单位盖章等。
该优化后的行政审批流程自2017年12月1日起开始实施。
为获得相关药品和医疗器械在我国上市许可,利用我国人类遗传资源开展国际合作临床试验的行政审批流程
一、适用范围
适用于为获得相关药品和医疗器械在我国上市许可,利用我国人类遗传资源开展国际合作的临床试验,其中药品临床试验包括I期、II期、III期以及生物等效性临床试验。
二、申请方式
由具有法人资格的合作双方,即中方单位和外方单位共同申请。其中,中方单位是指大陆境内的内资科研机构、高等学校、医疗机构、企业,外方单位是指外国组织及外国组织、个人设立的境内外机构,港、澳、台地区组织、企业或个人及设立的机构参照外方单位进行管理。合作发起方同临床机构协商决定填报主体。鼓励多中心临床研究设立组长单位,一次性申报。
三、审批条件
(一)基本条件。
1.采集、收集人类遗传资源的目的明确合法;
2.采集、收集计划方案合理;
3.具有负责人类遗传资源管理的部门和管理制度;
4.具有与采集、收集活动相适应的场所、设施、设备和人员;
5.经伦理委员会审查同意;
6.合作各方具有开展相关工作的基础和能力;
7.人类遗传资源提供者知情同意书格式文本规范;
8.人类遗传资源来源明确、合法,种类、数量与研究内容相符;
9.合作期限合理;
10.合作协议文本草案规范;
11.知识产权归属明确,分享方案合理;
12.对中国公众健康、国家安全、社会公共利益没有危害;
13.符合法律法规规定的其他条件。
(二)申请设立组长单位多中心临床研究国际合作事项的,伦理审查进行如下优化。
1.参与合作的临床机构认可组长单位伦理审查的,不需要再次进行伦理审查,仅需提供认可承诺书,依照组长单位行政许可审批决定即可开展国际合作事项;
2.参与合作的临床机构不认可组长单位伦理审查的,本单位伦理审查批件可在受理时与组长单位的同时提交,也可在项目获批后提交科技部,科技部确认后即可开展国际合作事项。
四、申请材料
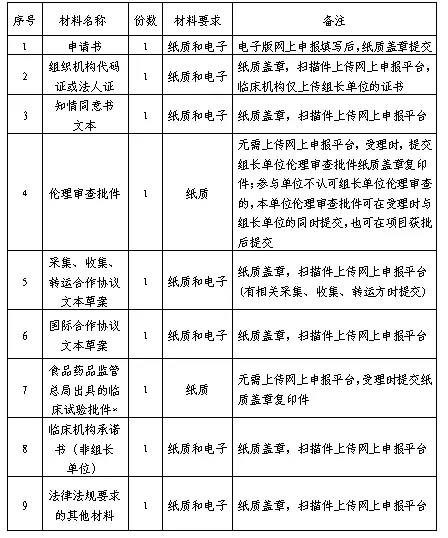
*备注:按照《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》有关精神,未获得临床试验批件,但受理临床试验申请达到规定期限后,食品药品监管部门未给出否定或质疑意见视为同意开展临床试验的,提供相关受理证明。
五、项目变更
在临床试验开展过程中,涉及人类遗传资源的合作方、研究目的、研究内容、合作期限等重大事项变更的,进行《人类遗传资源采集、收集、买卖、出口、出境审批》变更申报。
六、在线填报项目实施情况
填报单位按要求在项目实施过程中,组织合作单位在科技部网上监管系统平台填报项目进展情况。
七、权责分配
1.合作发起方对为获得相关药品和医疗器械在我国上市许可,利用我国人类遗传资源开展国际合作的临床试验负主要责任,须保证所申报材料的真实、完整、规范。在药物临床试验具体实施过程中发生重大研究方案变更时,合作发起方有责任主动同填报单位共同进行变更申报。
2.填报单位负责汇总国际合作项目相关文件,以及项目进展情况相关材料,按要求完成网上申报平台和监管系统平台相关信息的填报。
3.临床机构负责本单位承担的药物临床试验项目实施,提供伦理审查批件、知情同意文本、合作协议文本等申请相关文件,负责保护受试者的合法权益;设立组长单位的临床研究,组长单位负责药物临床试验项目启动,协助发起方协调各参与临床机构开展项目研究。
4.合同研究组织、第三方实验室等相关单位分别负责对本单位具体实施行为进行规范管理。
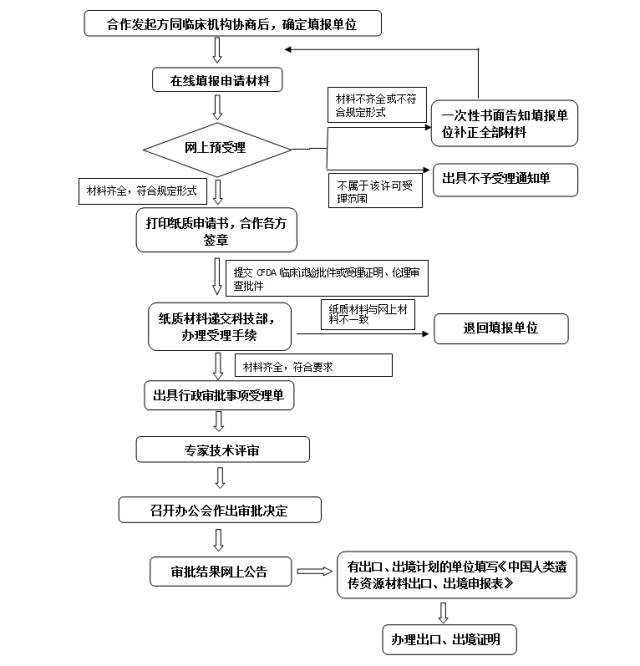
八、办理流程
全国服务电话:400-696-7707
客服电话:15003718880 / 15003718881
15003718885 / 15003718887( 同微信 )
咨询QQ:58775877 / 79538889

 在线咨询
在线咨询













